
测序数据质量控制
作者:网络转载 发布时间:[ 2016/1/13 14:29:09 ] 推荐标签:质量管理 软件测试管理
基于边合成边测序(Sequencing By Synthesis,SBS)技术,Illumina HiSeq2500高通量测序平台对cDNA文库进行测序,能够产出大量的高质量Reads,测序平台产出的这些Reads或碱基称为原始数据(Raw Data),其大部分碱基质量打分能达到或超过Q30。Raw Data通常以FASTQ格式提供,每个测序样品的Raw Data包括两个FASTQ文件,分别包含所有cDNA片段两端测定的Reads。
FASTQ格式文件示意图如下:

FASTQ格式文件示意图
注:FASTQ文件中通常每4行对应一个序列单元:第一行以@开头,后面接着序列标识(ID)以及其它可选的描述信息;第二行为碱基序列,即Reads;第三行以“+”开头,后面接着可选的描述信息;第四行为Reads每个碱基对应的质量打分编码,长度必须和Reads的序列长度相同。
测序碱基质量值
碱基质量值(Quality Score或Q-score)是碱基识别(Base Calling)出错的概率的整数映射。通常使用的Phred碱基质量值公式为:
公式中,P为碱基识别出错的概率。下表给出了碱基质量值与碱基识别出错的概率的对应关系:
表1 碱基质量值与碱基识别出错的概率的对应关系表
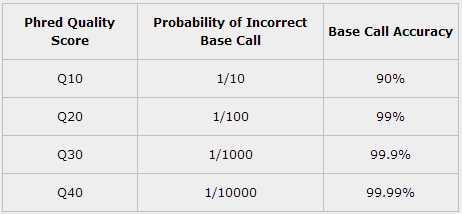
碱基质量值越高表明碱基识别越可靠,碱基测错的可能性越小。比如,对于碱基质量值为Q20的碱基识别,100个碱基中有1个会识别出错;对于碱基质量值为Q30的碱基识别,1,000个碱基中有1个会识别出错;Q40表示10,000个碱基中才有1个会识别出错。
以测序循环为单位,对单个样品所有Reads平行测序的碱基质量值做分布图,可以查看单个样品各个测序循环及整体的测序质量。
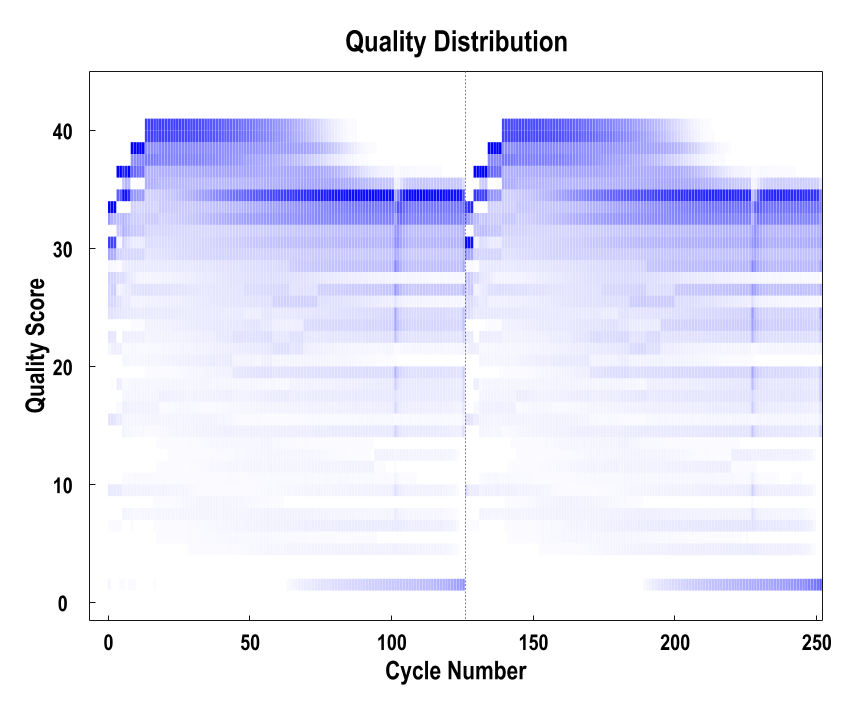
碱基质量值分布图
注:横坐标为测序碱基在Reads上的位置,纵坐标为碱基质量值。颜色深浅表示碱基比重,颜色越深,说明该位置测定的碱基中为对应质量值的碱基所占的比重越大,反之亦然。
测序质量控制
FASTQ文件中测序Reads需要与指定的参考基因组进行序列比对,定位cDNA片段在基因组或基因上的位置。在序列比对之前,首先需要确保这些Reads有足够高的质量,以保证后续分析的准确。测序质量控制方式如下:
(1) 去除测序接头以及引物序列;
(2) 过滤低质量值数据,确保数据质量。
经过上述一系列的质量控制之后得到的高质量Reads或碱基,称为Clean Data。Clean Data同样以FASTQ格式提供。
测序数据产出统计
某项目各样品数据产出统计见下表:
表2 样品测序数据评估统计表
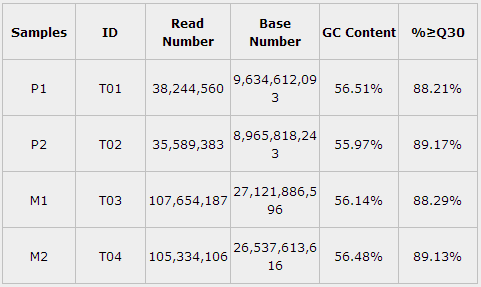
注:Samples:样品信息单样品名称;ID:样品编号;Read Number:Clean Data中pair-end Reads(双末端测序)总数;Base Number:Clean Data总碱基数;GC Content:Clean Data GC含量,即Clean Data中G和C两种碱基占总碱基的百分比;%≥Q30:Clean Data质量值大于或等于30的碱基所占的百分比。
转录组数据与参考基因组序列比对
获得Clean Reads后,将其与参考基因组进行序列比对,获取在参考基因组或基因上的位置信息,以及测序样品特有的序列特征信息。
TopHat2是一个高效的序列比对软件。它以高通量Reads比对软件Bowtie为基础,将转录组测序Reads比对到基因组上,然后通过分析比对结果识别外显子之间的剪接点(Splicing Junction)。这不仅为可变剪接分析提供了数据基础,还能够使更多的Reads比对到参考基因组,提高了测序数据的利用率。
转录组测序数据中,只有比对到参考基因组上的数据才能用于后续分析。因此,将比对到指定的参考基因组上的Reads称为Mapped Reads,对应的数据称为Mapped Data。
相关推荐











更新发布
功能测试和接口测试的区别
2023/3/23 14:23:39如何写好测试用例文档
2023/3/22 16:17:39常用的选择回归测试的方式有哪些?
2022/6/14 16:14:27测试流程中需要重点把关几个过程?
2021/10/18 15:37:44性能测试的七种方法
2021/9/17 15:19:29全链路压测优化思路
2021/9/14 15:42:25性能测试流程浅谈
2021/5/28 17:25:47常见的APP性能测试指标
2021/5/8 17:01:11
 sales@spasvo.com
sales@spasvo.com